Experimental Procedures
Cell Cultures
- Seed HBV stable cell lines, such as HepG2.2.15 (5), or tetracycline-inducible (tet-off) HepAD38 (6), or tetracycline-inducible (tet-off) HepDE19/DES19 cells (7, 8), in collagen coated 6-well plates at approximately 100% confluence with appropriate culture medium.
Note: Collagen-coated plate is ideal for cells to form confluent monolayers, especially for HepG2-based cells.
Note: HepG2.2.15 cell line is cultured with DMEM/F12 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin, plus 400 μg/mL G418. Tetracycline-inducible (tet-off) cell lines are maintained in the same way as the HepG2.2.15, but with the addition of 1 μg/mL tetracycline.
- When the cell monolayer reaches complete confluence (normally 24-48 hours post seeding), change the culture medium with fresh medium. For tetracycline-inducible cell lines, remove the tetracycline from culture medium to induce HBV replication.
Note: To obtain a high level of HBV replication in tetracycline-inducible cell lines, it is critical to withdraw the tetracycline when the cells reach confluence, as HBV replication will arrest cell growth.
Note: HBV infected cells can also be subjected to the following procedures.
cccDNA Extraction from Cell Culture
- Lyse cells by adding 1.5 mL of TE buffer (10:10) and 0.1 mL of 10% SDS into each well of a 6-well cell culture plate. Gently mix and incubate the plate for 30 min at room temperature.
- Transfer the viscous cell lysate to a 15 mL centrifuge tube. Add 0.4 mL of 5 M NaCl and gently invert the tube 10 times. Let the tube sit at 4°C for at least 16 h to efficiently precipitate proteins and protein-associated DNAs.
Note: Use wide-mouth plastic transfer pipette to transfer the lysate into centrifuge tube.
- Centrifuge at 14,500 × g for 30 min at 4°C. Transfer the supernatant to a fresh 15 mL centrifuge tube. To remove the residual protein from the supernatant, add an equal volume of phenol (approx. 2 mL) to the supernatant and mix thoroughly by hand shaking for 10 sec. Centrifuge at 3,500 × g for 10 min at 4°C, and transfer the aqueous phase to a fresh 15 mL tube. Repeat phenol extraction step. Add equal volume of phenol/chloroform (approx. 2 mL) and mix thoroughly by hand shaking, centrifuge at 3,500 × g for 10 min at 4°C and transfer the aqueous phase to a fresh 15 mL tube.
Note: Use leak-proof tube with screw cap. Do not shake the tube too vigorously or too long, which it may physically nick the supercoiled cccDNA.
- Add two volumes of 100% ethanol (approx. 4 mL) and mix thoroughly by inverting the tube several times. Incubate at room temperature overnight to precipitate DNA.
Note: Do not incubate at low temperature. Because high concentration of salt is used in Hirt extraction, incubation of DNA solution at low temperature will precipitate high amount of salt.
- On the third day, centrifuge the tube at 3,500× g for 30 min at 4°C, and discard the supernatant. Add an equal volume of 75% ethanol (approx. 2 mL) and gently flick the tube to wash the DNA pellet. Centrifuge at 3,500× g for 15 min at 4°C.
- Discard the supernatant. Allow the pellet to air dry for about 10 min at room temperature. Dissolve the DNA pellet in 25 μL TE buffer (10:1).
Agarose Gel Electrophoresis
- Prepare 150 mL of 1.2% gel by mixing 1.8 g agarose, 5 mL of 30× TAE buffer, and 145 mL of distilled water in a microwavable flask. Microwave the flask until the agarose is thoroughly dissolved. Let it cool down to about 50°C at room temperature. Pour the liquid gel into the gel tray (12cm×10cm) with an appropriate comb inserted. Let the gel solidify at room temperature before removal of the comb.
- Prepare 600 mL of gel running buffer by diluting 20 mL of 30× TAE buffer with de-ionized water.
- Prepare samples by adding approximately 3 μL of 10× loading buffer to 25 μL of each sample. Pipette up and down to mix well.
- Assemble submarine gel running unit by correctly positioning the gel box, adding running buffer into the chamber, and loading 28 μL of each sample into a separate well. Connect the gel running unit to a power supply and run gel at 25 V for overnight.
Southern Blot
- Disconnect the gel running unit and take out the gel carefully.
- Submerge the gel in a tray containing freshly prepared 0.2 M HCl. Agitate gently for 10 min at room temperature to depurinate the DNA samples from the gel. The bromophenol blue dye in the gel should gradually turn grey during this process.
- Rinse the gel three times with distilled water.
- Submerge the gel in a tray of denaturing buffer, and agitate gently for 1 h at room temperature. The loading dye in the gel should turn back to blue during this process.
- Rinse the gel three times with distilled water.
- Submerge the gel in a tray of neutralization buffer, and agitate gently for at least 1 h at room temperature.
- Soak the Hybond-XL membrane (12×10cm) in a tray with de-ionized water for 5 min at room temperature. Replace the water with 20× SSC and continue to agitate for 15 min.
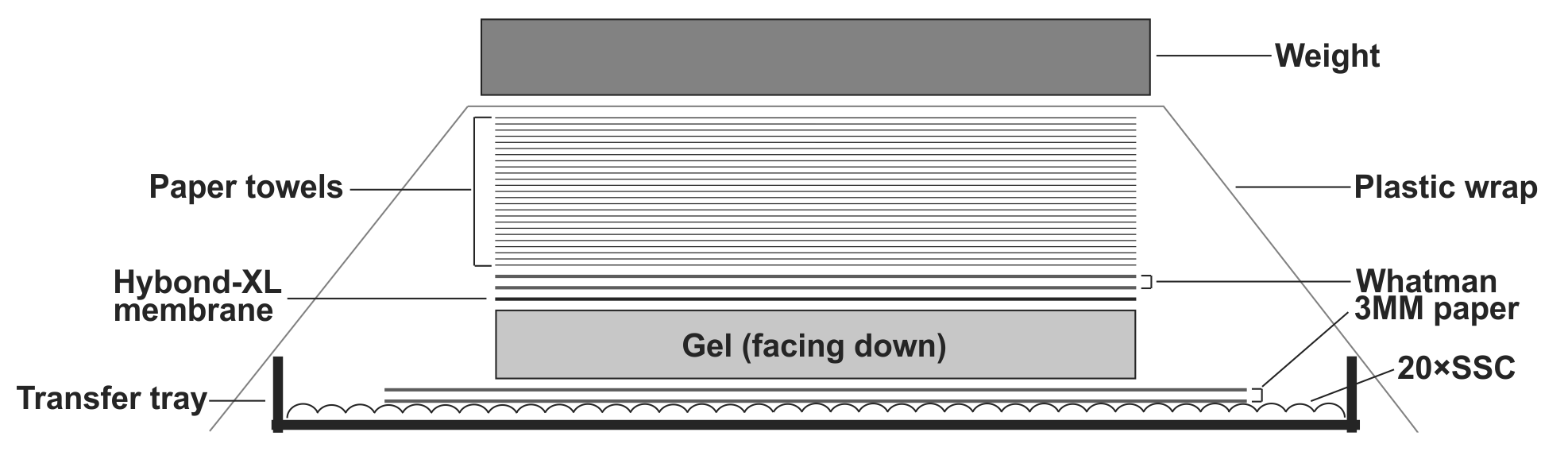
- To transfer DNA from the gel to the Hybond-XL membrane, assemble the transfer apparatus as following: layer two sheets of Whatman 3 MM filter paper in the gel transfer tray and pour 20× SSC transfer buffer on top to completely wet the Whatman papers. Smooth out the bubbles in between the Whatman papers and the tray gently by a plastic roller, and get rid of the excess amount of the transfer buffer. Place the gel on top of the Whatman papers with the side containing the loading wells facing down; make sure there is no air bubble between the Whatman papers and the gel. Place the presoaked Hybond-XL membrane on top of the gel and use a plastic roller to get rid of the air bubbles between the gel and the membrane. Layer two sheets of 20× SSC pre-wet Whatman papers (12×10cm) on top of the membrane, use plastic roller to remove air bubbles in between. Place a stack of dry paper towels (about 4 inches) pre-cut to the gel size on top of the Whatman papers. Seal the transfer tray with plastic wrap to prevent buffer evaporation during transfer. Put some weight (approx. 500g. such as metal plate, casserole dish etc.) on top of the transfer apparatus (Fig.1). Let the assembled transfer apparatus sit still on a flat surface and transfer for 24-48 h.
Note: When assemble the Southern blot transfer apparatus, avoid possible short-circuits of capillary liquid flow. The DNA will not be efficiently transferred if the blot short circuits. Use parafilm strips to seal the edges of the gel.
- After gel transfer, disassemble the blot apparatus and flip over the membrane with the gel still attached, and mark the position of each loading well on the membrane by a pencil. Wrap up the gel and paper towels by plastic wrap and discard.
Note: After transfer, the gel becomes thinner. Use the pencil tip to penetrate the loading wells and mark their positions on the membrane. This step is for labeling the DNA binding site of the membrane and tracking the running lanes with orientation.
- Crosslink the Hybond-XL membrane in a UV crosslinker chamber with UV energy dosage at 120 mJ/cm2. The membrane can be directly subject to hybridization, or sandwiched between two sheets of Whatman filters and stored at -20°C.
Probe Preparation for HBV DNA Detection
- Prepare rNTP (supplied with Riboprobe® system) solution by mixing 50 μL of each 10 mM rATP, 10 mM rGTP, 10 mM rCTP, and 0.2 mM UTP.
- Add 4 μL of 5 × reaction buffer, 4 μL of rNTP solution prepared from the previous step, 2 μL of 100 mM DTT, 0.25 μg of SalI-linearized pGEM-HBV DNA template, 1 μL of RNasin and 1 μL of SP6 polymerase into a nuclease free tube. Adjust the volume to 14 μL with nuclease-free water before adding 6 μL of [α-32P] UTP into the mixture. Incubate the reaction at 37ºC for 1 h.
Note: Linearization of the plasmid pGEM-HBV by SalI is an optimal step to produce “run-off” transcripts derived from the HBV sequence only. Other restriction enzyme, which singly cuts the vector sequences downstream of SP6-HBV transcription cassette but does not leave a 3’ overhang, can also be used.
Note: All radioactive materials and procedures should be handled strictly by following the institutional laboratory safety regulation rules.
- To remove the DNA template, add 1 μL of RNase-free RQ1 DNase to the reaction and incubate at 37°C for 15 min.
- Add 15 μL of 5M NH4OAc to stop the reaction. To precipitate the RNA probe, add 113 μL of nuclease-free water, 4 μL of yeast RNA, and 150 μL of isopropanol, gently mix and incubate at room temperature for 10 min.
- Centrifuge the mixture at 12,000× g for 30 min at 4°C, and discard the supernatant carefully without disturbing the pellet. Dissolve the probes in 400 μL of de-ionized formamide, followed by measurement of the counts per minute (CPM) of acid insoluble 32P with scintillation counter (PerkinElmer). Store the probes at -20°C.
Note: 32P-radiolabeled HBV DNA probes prepared by random priming or end labeling can also be used in the hybridization.
Hybridization
- Place the crosslinked membrane in a hybridization tube with the DNA-binding side facing the center of the tube. Add 5 mL of EKONO™ hybridization buffer, and pre-hybridize the membrane by rotating the hybridization tube at 65°C for 1 h in a hybridization oven.
Note: If using other commercial hybridization buffers, follow the pre-hybridization and hybridization conditions recommended by the manufacturers.
- Replace the pre-hybridization buffer with 5 mL of fresh EKONO™ hybridization buffer, and add HBV riboprobes with 1×107 Rotate the hybridization tube at 65°C overnight.
- Discard the hybridization solution on the following day, and wash the hybridization membrane with approximately half a tube of wash buffer. Rotate at 65°C for 30 min.
- Discard the wash buffer, and replace with half a tube of fresh wash buffer. Continue to wash at 65°C for 1 h.
- After the second wash, take out the membrane and dry it with paper towels, making sure the membrane is not cracked or wrinkled during this process. Seal the membrane with plastic wrap. Place the membrane in a phosphorimager cassette with the DNA-binding side facing up. Layer the phosphor imaging screen on top facing the DNA-binding side of the membrane. Close the cassette tightly and expose overnight in the dark.
- Scan the phosphor imaging screen with phosphorimager system. Store the membrane properly and erase the signal in the phosphor imaging screen with intense light for re-use.
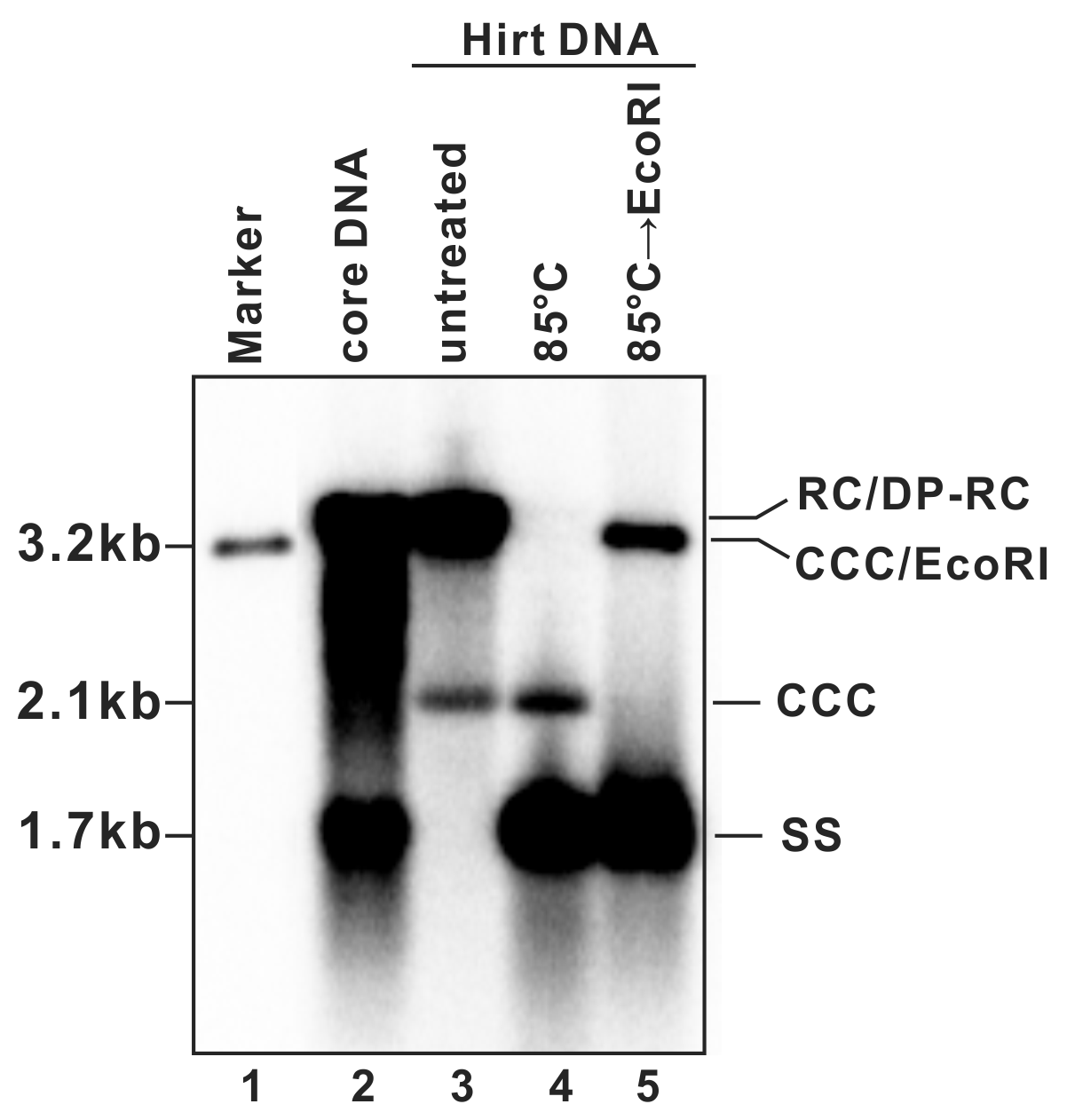
- A typical phosphor image of HBV DNA Southern blot is shown in Fig. 2 (also see in references (7, 9)). Quantify the signal intensity of the cccDNA bands with software provided by the phosphorimager system.
Figure 1: Assembly of HBV DNA Southern blot transfer apparatus. See text for details.
Figure 2: Detection of HBV DNA by a Southern blot assay. A typical pattern of HBV cytoplasmic core DNA (lane 2) and Hirt DNA (lane 3) upon Southern blot hybridization is shown. RC: relaxed circular DNA; DP-RC: deproteinized RC DNA; CCC: covalently closed circular DNA; SS: single-stranded (-) DNA. M: HBV genomic-length DNA marker. The approximate size of each HBV DNA species is labeled on the left. To further validate the authenticity of HBV cccDNA, the Hirt DNA sample in lane 3 has been heated to 85°C for 5 min before gel loading, a condition that denatures DP-rcDNA into SS DNA, while the cccDNA stays undenatured and its electrophoretic mobility remains unchanged (lane 4). The DNA sample from lane 4 is further digested with EcoRI, in which condition the cccDNA is linearized to a genome-length double-stranded DNA (lane 5). Reproduced from (
9) with permission from Elsevier.